اهمیت کُوِت در طیف سنجی
کوت یکی از اجزای دستگاه طیف سنج است که برای نگهداری نمونه استفاده میشود. بنابراین موقعیت قرارگیری و هندسه کوت روی دقت و صحت اندازه گیری جذب موثر است و باید به دقت کنترل شود.
برای انجام بهترین اندازه گیری، در بین اندازه گیری ها کوت باید درون نگهدارنده کووت باقی بماند. در صورت برداشته شدن، باید مراقب باشید که همیشه کوت را در همان حالت قبل در نگهدارنده کوت قرار دهید، برای این که این کار به درستی انجام شود، میتوانید از برچسب استفاده کنید تا جهت تابش نور از سمت منبع نور را علامتگذاری کنید. این کار اطمینان حاصل میکند که اثرات نوری برای اندازه گیری های مرجع و نمونه یکسان هستند.
برای دستیابی به بهترین نتیجه، اندازه گیری مرجع و نمونه باید با استفاده از همان نوع کوت انجام شود. کوت ها باید دارای پنجره های ساخته شده از مادهای باشند که در طیف مورد نظر شفاف باشد. برای دستیابی به بهترین نتیجه در اندازه گیری در محدوده UV ، باید از شیشههای شفاف در UV (UV-transparent glass) مانند شیشه کوارتز یا شیشه سوپراسیل (suprasil glass) استفاده شود.
کوت های یکبار مصرف، از پلی متیل متاکریلات (PMMA) ساخته شده اند، که این نوع کوتها در محدوده UV جذب دارند و مانند یک فیلتر (cut-off filter) عمل میکنند و باعث می شوند اندازه گیری جذب در محدوده UV صحت نداشته باشد و فقط باید برای اندازه گیری در محدوده مرئی قابل استفاده هستند.
در سامانههای ارزیابی مبتنی بر نور، مانند طیف سنجی فرابنفش-مرئی، تمیز بودنِ کوت ها تأثیر به سزایی در نتایج دارد. پنجره های کوت، که نور از آنها عبور میکند، باید قبل از هر استفاده با دستمال های بدون پرز (برای جلوگیری از خراش سطح توسط پرزهای دستمال) تمیز شود. در صورت وجود مواد یا اثرات روی پنجرههای کوت مانند اثر انگشت یا گریس، که در این سامانه اجزای جذب کننده اضافی به شمار میروند، در نهایت جذب بالاتری اندازه گیری میشود. با تمیزکاری کامل قبل و بعد از استفاده میتوان از این امر جلوگیری کرد. برای جلوگیری از دست زدن به پنجره ها پس از اتمام تمیز کردن کوت، باید مراقبت بیشتری انجام شود.
در نهایت، ذرات شناور درون کوت قابلیت انحراف پرتوی نور را دارند و منجر به جذبِ پس زمینه (background absorbance) میشوند. این انحراف نور به عنوان پراکندگی نور (light scattering) نیز شناخته می شود.
انتخاب حلال مناسب
حلالی که برای تهیه محلولهای طیف سنجی انتخاب میشود، باید در ناحیه طیف مورد نظر، شفاف باشند و قابلیت حل کردن مقدار کافی از نمونه را داشته باشد و بتواند محلول یکنواخت و خوبی تشکیل دهد؛ به طوری که ذرات حل نشده و نامحلول به صورت شناور وجود نداشته باشد.
هر حلالی که برای تهیه محلول به کار میرود، یک طول موج خاصی را جذب میکند؛ بنابراین موقعی که میخواهیم حلالی را انتخاب کنیم، مهم است که بدانیم طول موج جذب حل شونده در گستره طول موج جذب حلال نباشد.
نکته مهمی که وجود دارد این است که حلالی که برای تهیه محلول استفاده میشود، علاوه بر در نظر گرفتن تاثیر آن بر شفافیت محلول نهایی، باید تاثیرات آن بر جذبِ مادهی جاذب و در نتیجه شکل طیف جذب را هم بررسی کرد. به طور کلی حلالهای ناقطبی و مولکولهای ناقطبی حداقل تاثیر را بر روی یک دیگر دارند، در صورتی که مولکولهای قطبی هنگامی که با حلالهای قطبی برهمکنش دارند، اثرات نسبتا شدیدی در طیف جذبی دیده میشود.
برهمکنش بین حل شونده و حلال منجر به گسترش باند جذب و در نتیجه کاهش در وضوح ساختاری طیف جذبی (structural resolution) و حداکثر ضریب خاموشی (maximum extinction coefficient) میشود. این گونه برهمکنشها بین حلال و حلشونده، ساختارهای ارتعاشی را از بین میبرند. بنابراین در صورتی که تمایل داشته باشیم تا جزئیات طیفی را ببینیم، باید در استفاده از آنها اجتناب شود. حلالهای قطبی مثل آب، الکلها، استرها و کتونها باعث میشوند جزئیات ظریف پیکهای جذبی که ناشی از ارتعاشات است، در طیف جذبی نهایی دیده نشود (شکل۱).
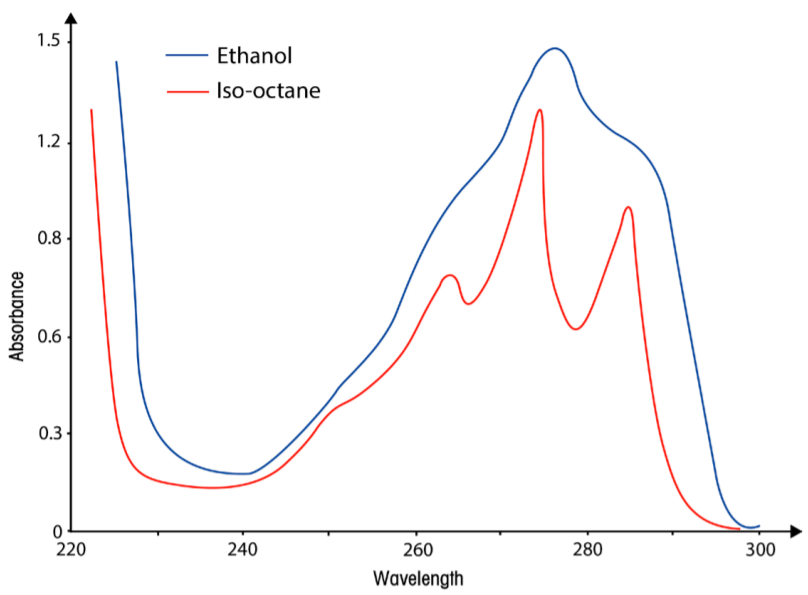
شکل ۱: اثر حلالهای قطبی و ناقطبی بر طیف جذبی فنول. محلول فنول در حلالهای اتانول (خط آبی) و ایزواکتان (خط قرمز).
در شکل ۱ طیف جذبی فنول را که در حلالهای اتانول و ایزواکتان حل شده است، دیده میشود. همانطور که مشخص است، طیف جذبی محلول فنول در اتانول (حلال قطبی) در مقایسه با محلول فنول در ایزواکتان (حلال ناقطبی) رزولوشون خوبی ندارد، به عبارت دیگر پیکهای کوچک طیف قابل تفکیک با یکدیگر نیستند، و این در حالتی است که محلول فنول در ایزواکتان جزئیات پیکهای جذب را در طیف جذبی بهتر نشان داده است. بنابراین برای دیدن پیکهایی نزدیک به پیکهای فاز گازی ماده، از محلولهایی استفاده میکنند که حلالشان ناقطبی باشد (شکل ۲). حلالها ناقطبی رایج برای طیف سنجی جذبی، هیدروکربنها هستند. در شکل ۲ هم مشابه شکل ۱ دیده میشود که پیکهای جذبی کوچک ماده در طیف مربوط به محلول اتانول قابل تفکیک نیستند.
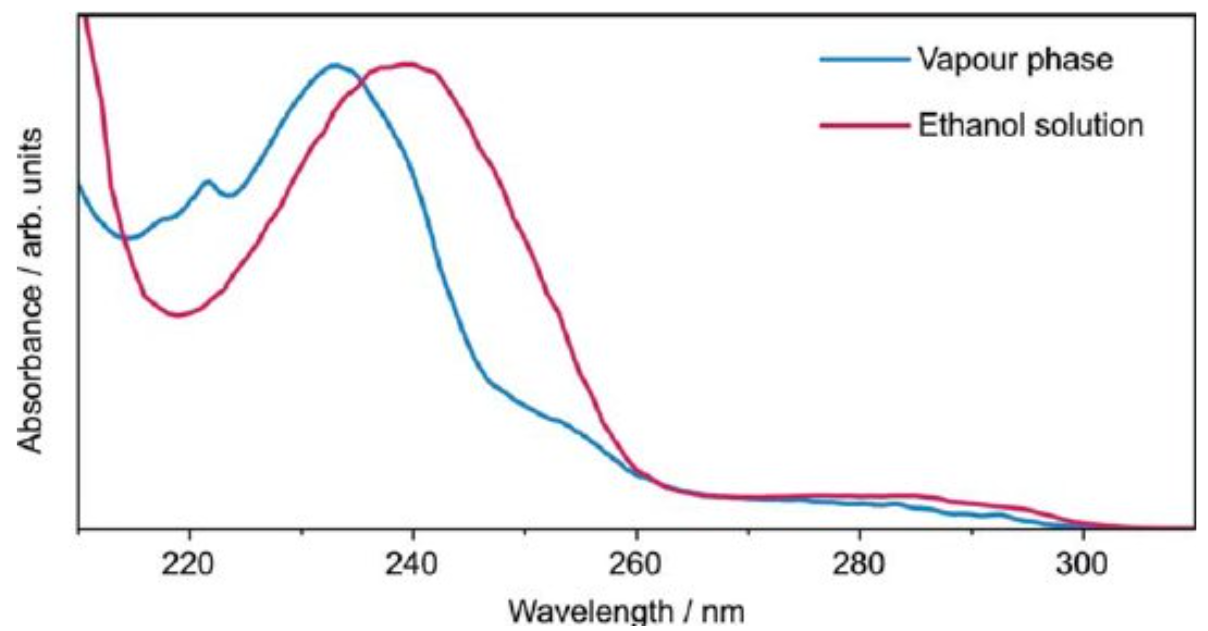
شکل ۲: طیف جذب فرابنفش p -MePhSH در فاز گاز (خط آبی) و در محلول اتانول (خط قرمز).
موقعیت پیکهای جذبی تا حد زیادی تحت تاثیر ماهیت حلال قرار میگیرد (در شکل ۲ به وضوح مشخص است.). به طور مثال یک نوار جذب ضعیف در ناحیه ۲۸۰ نانومتر تا ۲۹۰ نانومتر که در اثر افزایش قطبیت حلال به طرف طول موجهای کوتاهتر جا به جا شود، نشانهی قاطعی برای حضور کربونیل است. همچنین یک نوار جذب ضعیف در حدود ۲۶۰ نانومتر با نشانههایی از ساختار ظریف ارتعاشی، نشاندهنده وجود حلقه آروماتیک است.
حلالهای رایج برای طیف سنجی در ناحیه فرابنفش طیف، عبارتند از آب، اتانول ۹۵% و سیکلوهگزان و برای طیف سنجی در ناحیه مرئیِ طیف، هر حلال بی رنگی که در ناحیه مرئی جذب نداشته باشد، مناسب است. هنگام مقایسه طیفهای جذبی با نمونه استاندارد با هدف شناسایی، باید حلال یکسانی استفاده شود.
انتخاب طول موج مناسب
برای رسیدن به بیشینه حساسیت، اندازهگیریهای طیف سنجی جذب مواد، باید در طول موجی انجام شود که مربوط به پیک جذب آن ماده است؛ زیرا در این طول موج، جذب به ازای واحد غلظت، بزرگترین تغییرات را از خود نشان میدهد و بنابراین حداکثر دقت را ایجاد میکند. به علاوه منحنی جذب اغلب در این ناحیه مسطح است و تحت این شرایط میتوان انتظار داشت که قانون بیر-لامبرت صادق باشد.
اهمیت اندازهگیری جذب مواد به طور دقیق در یک طول موج خاص که همان لاندای ماکسیموم (lambda max) طیف جذب است، در شکل ۱ نشان داده شده است. در شکل ۱ تنظیم طول موج به صورت یک نوار طیفیِ باریکِ در نزدیکی طول موج جذب بیشینه مثل نوار طیفی A، تأثیر محسوسی بر میزان جذب در پیک جذب نخواهد داشت، در حالی که اگر نوار طیفی با پهنای مشابه در نواحی طیف طول موجهای کمتر تنظیم شود مثل نوار طیفی B، بسته به نقطهی دقیق درون نوار طیف که در آن اندازهگیری انجام میشود، احتمال ایجاد خطای اساسی در طیف وجود خواهد داشت. به عبارت دیگر، در صورتی که طیف انتخابی برای بررسی طیف سنجی جذبی یک ماده در بیشینه طول موج جذب باشد، به ازای تغییر کم طول موج درون طیف تابشی، تغییر جذب محسوس نخواهد بود، و همین تغییر جذب نامحسوس در اثر تابشی طیف کوچکی از طول موجها، منجر به نتایج دقیق تری برای بررسی غلظت محلول میشود. در این حالت است که رابطه خطی بیر-لامبرت برای محاسبه غلظت صادق خواهد بود. همانطور که در شکل ۳ دیده میشود، در صورتی که طیف تابشی در دستگاه طیف سنج، از ناحیه بیشینه جذب آن ماده فاصله داشته باشد، در اثر تغییر طول موج تابشی در آن طیف طول موجی، میزان جذب بسیار تغییر میکند و همین باعث میشود که سنجیدن غلظت ماده از طریق رابطه بیر-لامبرت، اعتبار نداشته باشد.
خطاهای اندازهگیری ناشی از تنظیم طول موج یا کالیبراسیون دستگاه در صورتی که اندازهگیری در بیشینه طول موج جذب (یا همان lambda max) انجام شود، کمینه است.
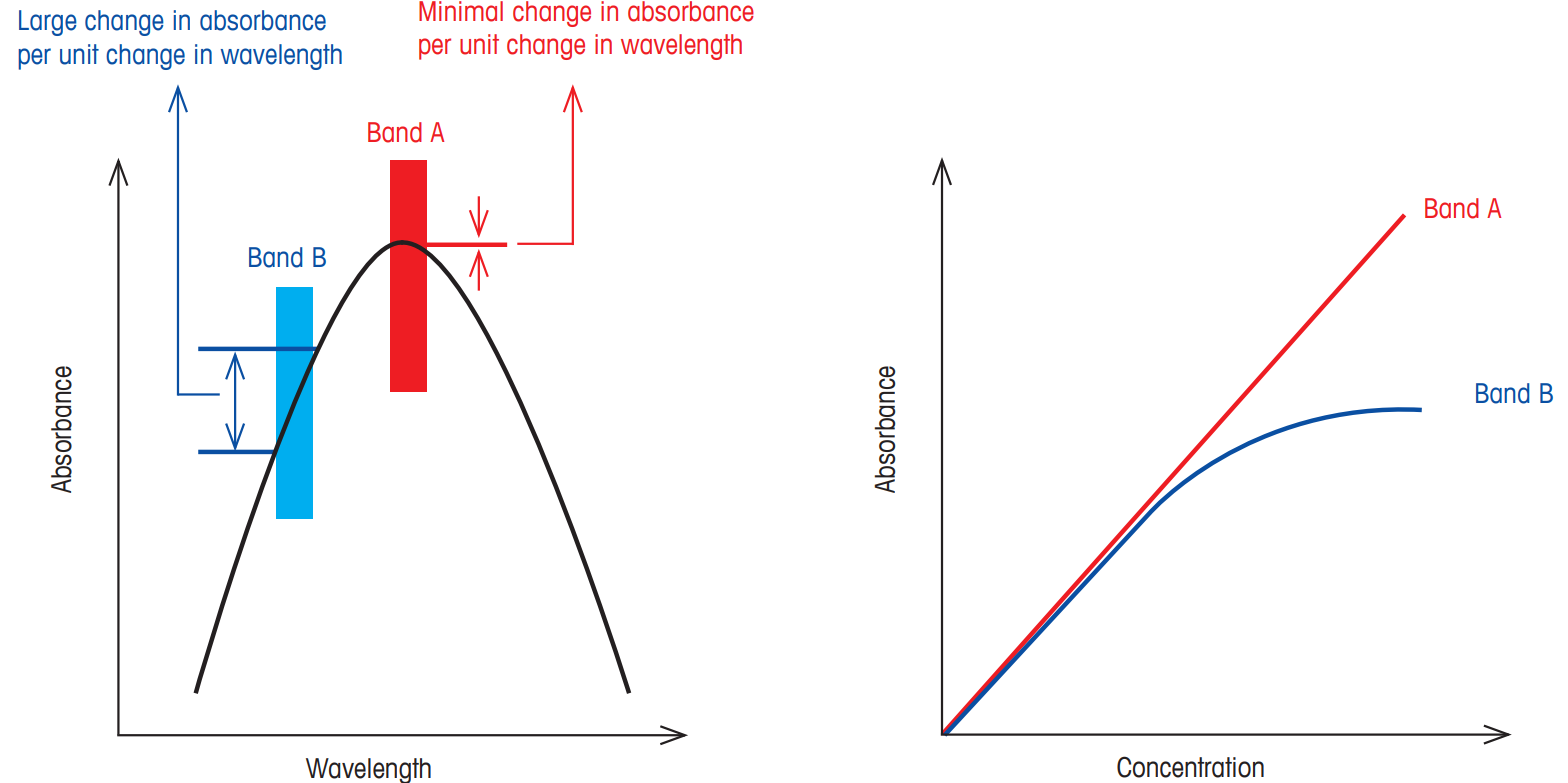
شکل ۳: تصویر سمت چپ: تغییر جذب بر حسب تغییر طول موج تاثیر محسوسی در باند B دارد؛ در حالی که در باند A میزان تغییر جذب بسیار کم و قابل صرف نظر کردن است. تصویر سمت راست: این تغییر بر غلظت محاسبه شده در حالت B موثر است.
غلظت نمونه
برای حصول نتایج بهینه ناشی از طیف سنجی که با قانون بیر-لامبرت تطابق داشته باشد، میزان جذب باید در محدوده خطی دستگاه تعیین شود. بنابراین، بهتر است در طیف سنجی نمونه از مقادیر جذب بسیار بالا (بیشتر از ۲.۵) و جذب بسیار کم (کمتر از ۰.۳) که ممکن است منجر به غیر خطی بودن شود، خودداری شود. با این وجود، برخلاف سایر تکنیک های تحلیلی، نمونههایی با غلظت بسیار کم مانند نمونههایی با غلظت ۰.۰۱ مولار را میتوان با استفاده از طیف سنجی فرابنفش-مرئی مشخصهیابی کرد. یک نمونه معمول از این نوع محلولها زمانی است که میخواهند کیفیت آب را بررسی کنند، دیده میشود که در آن باید غلظت بسیار کم مواد تعیین شود.
وقتی یک ماده آنالیت (analyte) برهمکنش دارد (ترکیب یا تفکیک) و یا با حلال واکنش وارد واکنش میشود، گونههای شیمیایی جدیدی به وجود میآیند که منجر به تغییر رفتار جذب نمونه میشود. بنابراین توصیه میشود که برای جلوگیری از هرگونه واکنش جانبی، از یک حلال پایدار (a non-interacting solvent) و آنالیت با غلظت کم استفاده کنید.
برای درک بهتر مطالب در بخش «غلظت نمونه» و «انتخاب طول موج مناسب»، در بخش بعد به تعیین غلظت نمونههای طیف سنجی به طور تجربی پرداخته شده است.
تعیین غلظت نمونه از طریق کمی سازی
کمی سازی یا تعیین غلظت یک نمونه از طریق طیف سنجی فرابنفش-مرئی مبتنی بر قانون بیر-لامبرت است. این قانون بیان میکند که جذب یک محلول به طور مستقیم با غلظت مادهی جاذب در محلول و ضخامت کُوت (به عبارت دیگر، طول مسیر عبور نور) متناسب است. بنابراین وقتی که ضخامت کُوت را ثابت در نظر بگیریم، طیف سنجی فرابنفس-مرئی میتواند برای تعیین غلظت ماده جاذب درون محلول استفاده شود. اگرچه، این مهم است که بدانیم میزان جذب در اثر تغییر غلظت چقدر تغییر میکند. این مورد را میتوانیم از مراجع به دست بیاوریم، مقل جداول ضریب جذب (extinction coefficients) یا راه خیلی ساده تر استفاده از منحنی کالیبراسیون است. به طور مثال، برای نمونهی ناشناس (نمونهای که ندانیم چه چیزی است)، باید از منحنی کالیبراسیون استفاده کرد.
اولین گام در آنالیز کمی انتخاب طول موج مناسب است که طبق بخش «انتخاب طول موج مناسب» متوجه شدیم که باید در محدوده طول موج بیشینه جذب باشد تا حساسیت و دقت لازم برای اندازه گیری وجود داشته باشد. اگر نوار طیف طول موج بیشینه انتخاب شود، اثر نسبی سایر مواد موجود در محلول و ناخالصیها کمتر در اندازه گیریها موثر خواهد بود. به علاوه نرخ تغییر جذب با طول موج کوچکتر است و اندازه گیری خیلی تحت تاثیر خطاهای کوچک قرار نمیگیرد.
گام بعدی کمی سازی، اندازهگیری استانداردهای شناخته شده در آن طول موج انتخابی است. بعد از اندازهگیری باید منحنی جذب نمونه بر حسب غلظت رسم شود. در شکل ۴ نمونهای از این منحنیها رسم شده است. با برازش خطی دادههای حاصل از اندازه گیری منحنی کالیبراسیون برای این نمونه حاصل شده است. در حالت ایده آل، حداقل به سه غلظت مختلف ماده نیاز است، اگرچه میتوان یک غلظت را نیز استفاده کرد. در عمل، استفاده از پنج غلظت مختلف منحنی کالیبراسیون دقیق تری ایجاد میکند. طبق رابطه بیر-لامبرت جذب با غلظت یک رابطه خطی دارد و نتایج حاصل از اندازه گیری را از طریق برازش مرتبه اول یا برازش خطی میتوان به صورت یک منحنی نشان قرار داد.
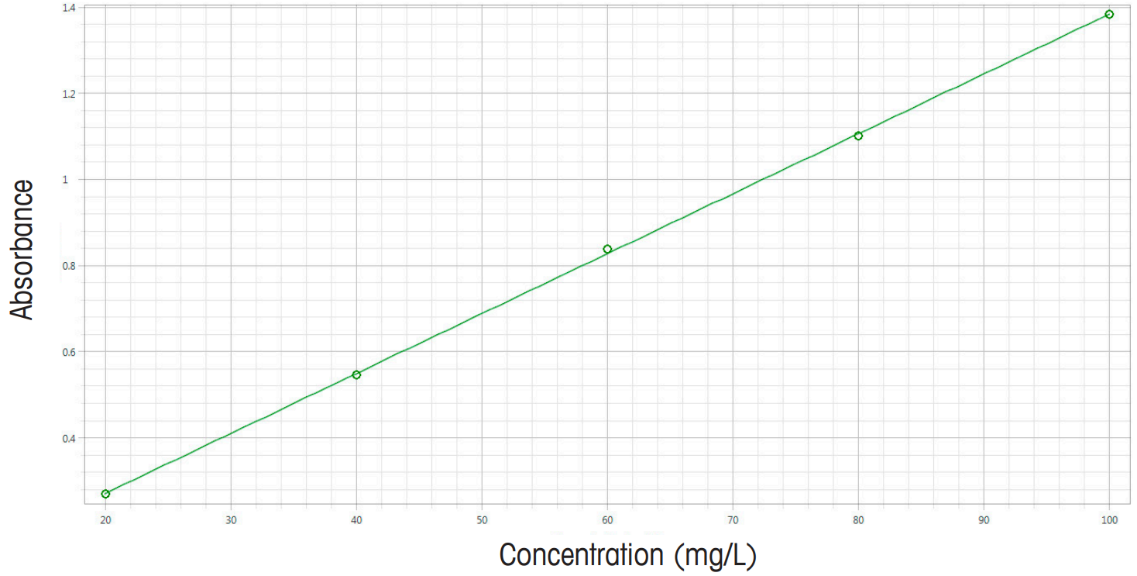
شکل ۴: مقادیر جذب یک محلول استاندارد بر حسب غلظت. برازش خطی (رگرسیون خطی) نقاط اندازهگیری شده، خط کالیبراسیون را میدهد.